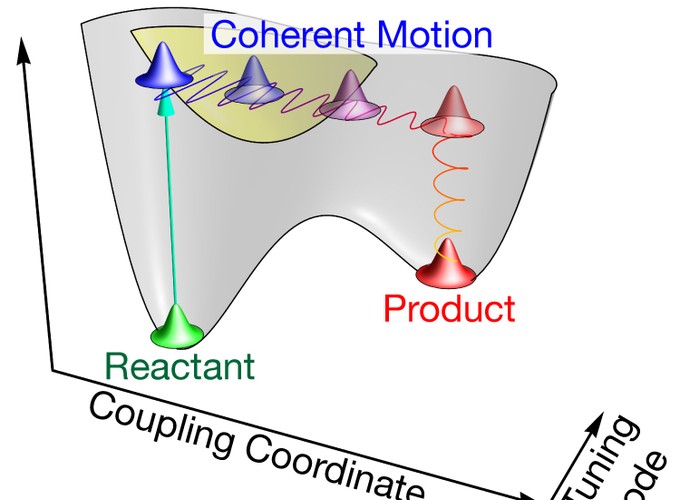
Femtosecond spectroscopy has revealed coherent wave packet motion time and time again, but the question as to whether these coherences are necessary for reactivity or merely a consequence of the experiment has remained open. For diatomic systems in the gas phase, such as sodium iodide, the dimensionality of the system requires coordinated atomic motion along the reaction coordinate. Coherent dynamics are also readily observed in condensed-phase multidimensional systems such as chromophores in proteins and solvated charge transfer dimers. Is precisely choreographed nuclear motion (i.e., coherence) required for reactivity in these systems? Can this coherence reveal anything about the reaction coordinate?
In this Account, we describe our efforts to tackle these questions using femtosecond stimulated Raman spectroscopy (FSRS). Results of four exemplary systems are summarized to illustrate the role coherence can play in condensed-phase reactivity, the exploitation of vibrational coherence to measure vibrational anharmonicities, and the development of two-dimensional FSRS (2D-FSRS). We begin with rhodopsin, the protein responsible for vertebrate vision. The rhodopsin photoreaction is preternaturally fast: ground-state photoproduct is formed in less than 200 fs. However, the reactively important hydrogen out-of-plane motions as well as various torsions and stretches remain vibrationally coherent long after the reaction is complete, indicating that vibrational coherence can and does survive reactive internal conversion. Both the ultrashort time scale of the reaction and the observed vibrational coherence indicate that the reaction in rhodopsin is a vibrationally coherent process. Next we examine the functional excited-state proton transfer (ESPT) reaction of green fluorescent protein. Oscillations in the phenoxy C–O and imidazolinone C=N stretches in the FSRS spectrum indicated strong anharmonic coupling to a low-frequency phenyl wagging mode that gates the ESPT reaction. In this case, the coherence revealed not only itself but also the mode coupling that is necessary for reactivity. Curious as to whether vibrational coherence is a common phenomenon, we examined two simpler photochemical systems. FSRS studies of the charge transfer dimer tetramethylbenzene:tetracyanoquinodimethane revealed many vibrational oscillations with high signal-to-noise ratio that allowed us to develop a 2D-FSRS technique to quantitatively measure anharmonic vibrational coupling for many modes within a reacting excited state. Armed with this technique, we turned our attention to a bond-breaking reaction, the cycloreversion of a cyclohexadiene derivative. By means of 2D-FSRS, the vibrational composition of the excited-state transition state and therefore the reaction coordinate was revealed.
In aggregate, these FSRS measurements demonstrate that vibrational coherences persist for many picoseconds in condensed phases at room temperature and can survive reactive internal conversion. Moreover, these coherences can be leveraged to reveal quantitative anharmonic couplings between a molecule’s normal modes in the excited state. These anharmonic couplings are the key to determining how normal modes combine to form a reaction coordinate. It is becoming clear that condensed-phase photochemical reactions that occur in 10 ps or less require coordinated, coherent nuclear motion for efficient reactive internal conversion.